
by Kaveen Tennakoon, PhD Student in Chemistry
19 August 2025
In the last decade, artificial intelligence (AI) has gone from being a futuristic buzzword to a practical, transformative force in the world of chemistry. No longer the exclusive domain of computer scientists, AI has rapidly become a must-have tool for chemists, reshaping how discoveries are made, data are analyzed, and experiments are designed. This three-part series takes a deep dive into the AI-driven revolution sweeping across chemistry, exploring breakthroughs in synthesis, materials discovery, advanced data analysis, and the AI tools making all of this possible. In this first part of our series on the AI revolution in chemistry, we’ll explore how machine learning and other AI technologies are propelling everything from organic synthesis to reaction mechanism discovery, and why their impact is being felt in every corner of chemical research.
AI in Synthesis: Planning Reactions and Uncovering Mechanisms
In organic chemistry, one of AI’s most striking impacts is on reaction planning and synthesis. Traditionally, chemists spent days or weeks devising synthetic routes to target molecules. Now, AI-driven retrosynthesis tools can propose viable pathways in minutes, searching reaction databases far beyond any single chemist’s memory. For example, Synthia platform (formerly Chematica) combines machine learning with expert-encoded reaction rules to drastically cut down route planning time “from weeks to minutes,” while still providing realistic, lab-ready pathways. Likewise, IBM’s RXN for Chemistry uses transformer neural networks trained on millions of reactions to predict reaction outcomes and suggest synthetic routes with over 90% accuracy. Chemists worldwide can access IBM RXN via a cloud interface, integrating it into their workflow without specialized hardware. These AI systems not only find conventional routes faster but often suggest novel disconnections or creative synthetic strategies that humans might overlook. Notably, the original Chematica system even helped reduce a complex drug synthesis from 12 steps to just 3 in one case, dramatically cutting cost and effort.
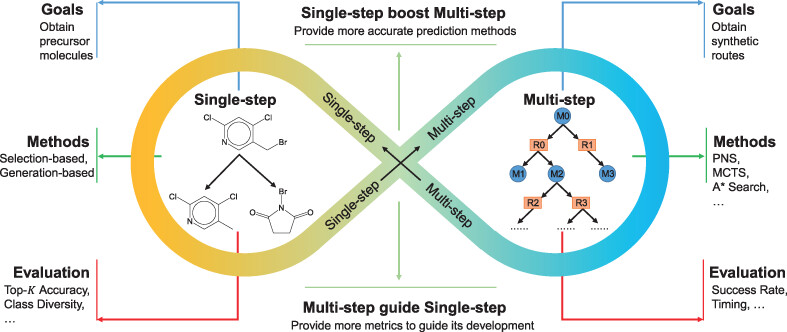
Beyond planning how to make molecules, AI is improving understanding of how reactions work. In 2023, researchers demonstrated that deep neural networks could analyze kinetic data and automatically identify the likely mechanism class of an organic reaction. In one study, a trained model correctly classified a wide range of catalytic mechanisms (even ones with tricky features like catalyst activation steps) and proved robust even when given sparse or noisy data. Such AI-guided mechanism classification can “streamline and automate mechanistic elucidation”, reducing reliance on tedious manual rate-law derivations. This means chemists can more quickly determine why a reaction yielded a specific outcome, rather than just predict what will happen. From suggesting synthetic routes to deciphering reaction pathways, AI is becoming a true collaborator to the synthetic chemist.
Machine Learning Accelerates Drug Discovery
Perhaps the most publicized impact of AI in chemistry has been in drug discovery. Designing a new drug is like finding a needle in a molecular haystack: chemists must search through countless possible molecules to find ones that bind a disease target, have suitable properties, and can actually be made. AI is fundamentally changing this search process. Modern machine learning models can predict a molecule’s properties (like biological activity, toxicity, or solubility) with impressive accuracy, helping researchers triage huge libraries of compounds in silico. Instead of physically testing thousands of molecules, scientists can use models to quickly narrow the field to a few promising candidates, saving enormous time and cost. For instance, the open-source Chemprop tool uses graph neural networks to predict molecular properties and has become a popular choice for academic groups to build QSAR models (quantitative structure–activity relationships). Another widely used library is DeepChem, which provides a rich collection of models and datasets to “democratize deep learning in drug discovery, materials science, chemistry, and biology”. With such tools, even researchers without a computer science background can train neural networks to predict activities of new compounds or scan for potential drug hits.
AI is also helping generate novel drug candidates. Generative models (like variational autoencoders and generative adversarial networks) can invent molecular structures with desired properties, rather than relying on human imagination alone. These models learn the patterns of what makes a molecule drug-like, then propose new candidates that fit the criteria, some of which may be unlike any existing compounds. In pharmaceutical companies and startups, generative AI systems have been designed molecules that went from computer to clinic in record time. In fact, the first AI-designed drug candidates entered human clinical trials around 2020, marking a pivotal milestone for the field. Since then, multiple AI-designed molecules (for example, by companies like Exscientia and Insilico Medicine) have advanced into Phase I trials, with several showing promising results. These early successes demonstrate how AI can significantly accelerate the pipeline: one AI-designed drug for fibrosis reached Phase I in under two years, roughly half the typical timeline.
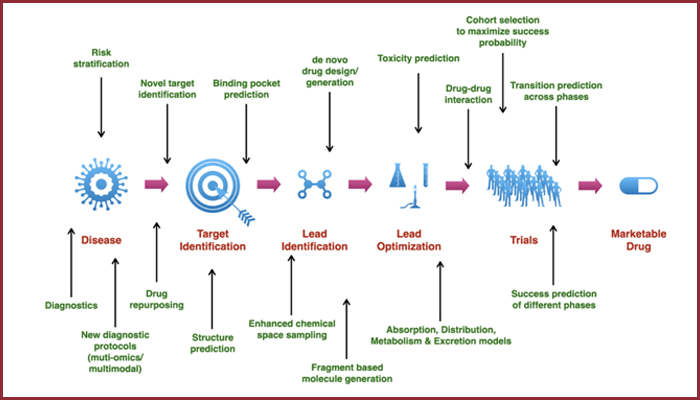
Importantly, AI in drug discovery isn’t just about finding a potent molecule; it’s also about balancing the many factors that make a successful drug. Modern algorithms consider synthetic accessibility and cost alongside biological activity. A recent MIT-led project, for example, created an AI framework called SPARROW to automatically choose molecule sets that maximize desired properties while minimizing the cost and complexity of synthesizing them. By integrating predictive models for activity with retrosynthesis planning tools, such approaches ensure that promising drug leads are not only potent but also feasible to make. This holistic, AI-driven strategy helps drug hunters avoid dead-ends and focus on candidates that are worth the effort. Combined with ever-improving virtual screening (often enhanced by physics-based simulation software such as Schrödinger’s suite), AI-driven workflows are significantly reducing the time required for drug discovery. The ultimate beneficiaries will be patients, as vital new medicines make it to the clinic faster and at lower cost.
From AI-powered retrosynthesis to automated reaction mechanism discovery, artificial intelligence is already transforming how chemists approach synthesis and drug development. But the revolution doesn’t stop at the molecular level. In Part 2, we’ll explore how AI is speeding up the search for next-generation materials powering self-driving labs, enabling autonomous experiments, and offering new analytical insights that are reshaping physical chemistry and materials science. Stay tuned to discover how the combination of machine learning and automation is moving the chemical sciences into an exciting new era.
Ready to put AI to work in your lab? Explore our curated AI tools for STEM and Literature Review at: Resource Center